The current version of SeqWare is 1.1.1, released on May 1st, 2015. See the release notes for details.
About
History
The SeqWare project (originally SolexaTools) was started in 2007 by Brian O’Connor while he was a postdoc in the Nelson Lab at UCLA.
Purpose
The open source SeqWare project is a portable software infrastructure designed to analyze massive genomics datasets produced by contemporary and emerging technologies, in particular Next Generation Sequencing (NGS) platforms. It consists of a comprehensive infrastructure focused on enabling the automated, end-to-end analysis of sequence data – from from raw base calling to analyzed variants ready for interpretation by users. SeqWare is tool agnostic, it is a framework for building analysis workflows and does not provide specific implementations out-of-the-box. You use SeqWare to create high-throughput infrastructure for NGS analysis using whatever analysis tools you like.
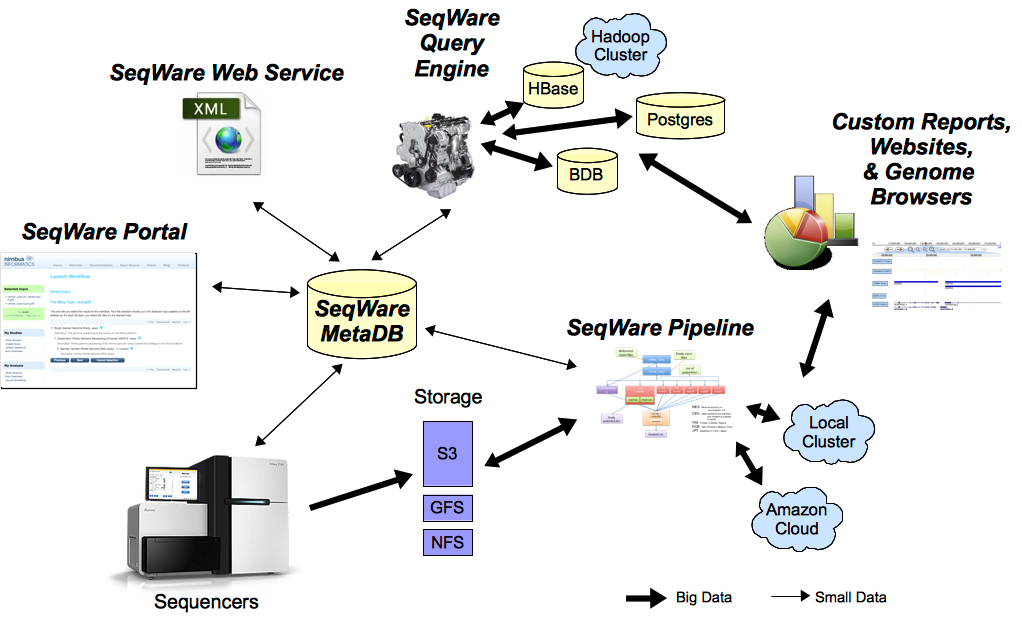
Components
SeqWare currently provides 5 main tools specifically designed to support massively parallel sequencing technologies. All tools can be used together or separately:
- MetaDB: provides a common database to store metadata used by all components.
- Portal: a LIMS-like web application to manage samples, record computational events, and present results back to end users.
- Pipeline: a workflow engine that is capable of wrapping and combining other tools (BFAST, BWA, SAMtools, etc) into complex pipelines, recording metadata about the analysis, and facilitates automation of pipelines based on metadata.
- Web Service: a programmatic API that lets people build new tools on top of the project
- Query Engine: a NoSQL database designed to store and query variants and other events inferred from sequence data.
Publications
Please cite the SeqWare paper:
O’Connor, B. D., B. Merriman, and S. F. Nelson. SeqWare Query Engine: storing and searching sequence data in the cloud. BMC Bioinformatics 2010, 11(Suppl 12):S2