SeqWare MetaDB
Overview
The SeqWare MetaDB is a metadatabase modeled on the Short Read Archive schema and built with PostgreSQL. It includes tables to model the run of the sequencers, analysis “processing” events, and installed workflows. A common database for tracking metadata, used by the other SeqWare components.
Building and Installing
Most users will want to use our pre-configured VMs, see the SeqWare Install Guide for how to get the VM. For those interested in installing the MetaDB from scratch please see the Install Guide
Tables
The SeqWare MetaDB was built on top of the Short Read Archive (SRA) data model. It is not a 100% translation of the data model, there may be places where the SRA model provides greater flexibility and places where we simplified the model for our own purposes. But we generally tried to keep with the SRA model as much as possible. You can find out more information from the following documents:
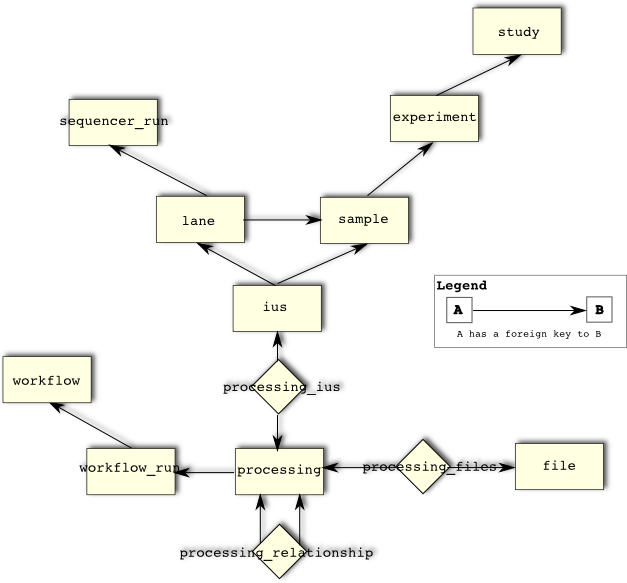
Sequence Data
SeqWare uses the MetaDB to track processing events and to connect samples to processed data and/or to connect one set of processed data to another. For example linking between a biological sample, a sequencing run, the generated raw sequence data, and downstream processed sequence data is possible. This is actually a complicated process that depends on the specific path through a pipeline, with details like multiple samples being pooled for sequencing, or lanes contain data from multiple samples (multiplexed or so-called bar-coded samples) arising. First, a simple example.
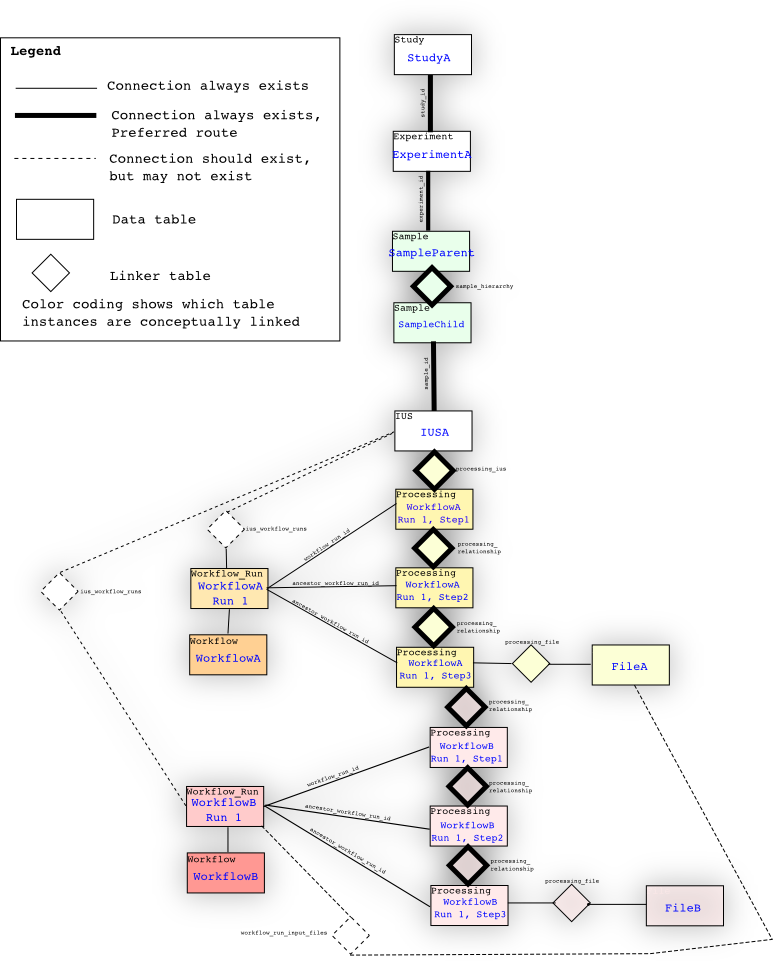
The ‘proper’ way to traverse the database from a study or sequencer_run is to go through ius, processing_ius, and processing. So from study, you traverse through experiment, sample to sample through the sample_hierarchy table, and ius, to processing_ius, and then to all of the processings. The processing tree starts from the processing linked through processing_ius, and then through the processing_relationship table. Each new processing event has the parent of the previous processing event; for example, if WorkflowA is run on IUSA, and then WorkflowB is also run on IUSA using the results from WorkflowA, the processing tree looks like IUSA -> processing_ius -> Processing of step 1,2,3 of WorkflowA -> Processing of step 1,2,3 of WorkflowB. Processings are linked to workflow_runs, which have connections to IUSes through ius_workflow_runs and/or Lanes through lane_workflow_runs, as a convenience only. These links are not necessarily present (though we try to make sure they are). The safest way to find all of the processing events linked with a Study is to traverse through the IUS to the root Processing through processing_ius, not through ius_workflow_runs.
The following is a more complicated database diagram that includes tables used for linking metadata to workflow runs and all attribute tables. This should be more useful for developers rather than users.
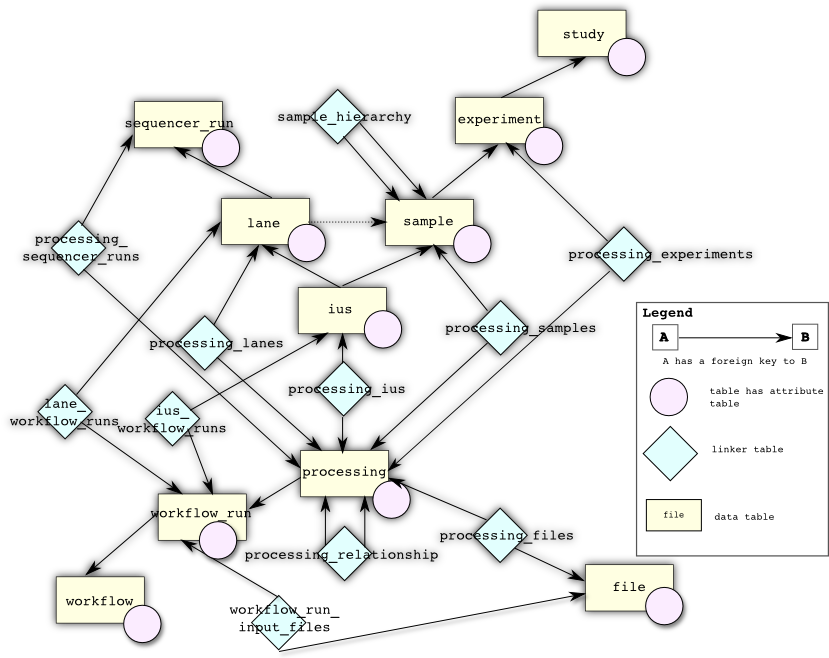
There is one large division between the tables at the “top” of the database (sequencer_run, lane, sample, ius, experiment, and study) which have physical counterparts and are typically created by migration by the LIMS and the tables at the “bottom” of the database (workflow, workflow_run, processing, and file) which are actually used by SeqWare to maintain details on what workflows have been installed and run.
Note there are also attribute tables for each of the above tables (of the form X_attribute) which maintain arbitrary key value information that can be used to guide custom behaviour at your particular SeqWare site.
Finally, there are a host of linking tables (of the form processing_X) which maintain information on which steps in your workflow are attributable to what physical entities.
For more in-depth information on our schema including table-by-table descriptions, please refer to this
Upgrading From 0.13.6.X to 1.0.X
First, let’s review which tools depend on what.
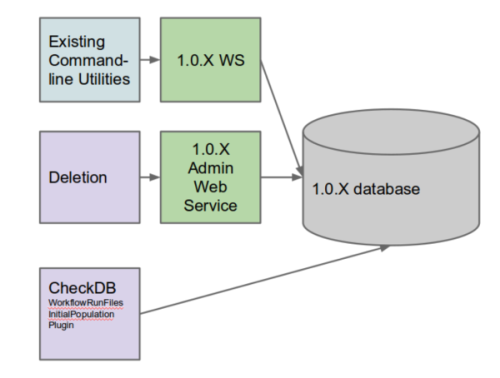
All currently supported command-line utilities from 0.13.6.X use the SeqWare web service and in their 1.0.X incarnation, will rely upon a 1.0.X web service. The new deletion utility relies upon the 1.0.X admin web service (to be documented). The checkDB and migration plugin (used to copy input file paths from the old workflow run reports) rely upon a direct database connection as well. Please refer to User Configuration for additional information on setting these up.
You will need both your SeqWare webservice and SeqWare database settings configured before attempting to update your database. The procedure is as follows:
First, run the various update scripts:
psql -U seqware seqware_meta_db < 0.13.6.x_to_1.0.1.sql
psql -U seqware seqware_meta_db < 1.0.1_to_1.0.3.sql
psql -U seqware seqware_meta_db < 1.0.4_to_1.0.5.sql
Second, run the migration plugin:
java -jar seqware-distribution/target/seqware-distribution-1.1.0-full.jar -p net.sourceforge.seqware.pipeline.plugins.WorkflowRunFilesInitialPopulationPlugin
Third, you’ll probably want to create the initial file-provenance-report (note, you’ll probably want this to run on a schedule for updates)
seqware files refresh
Optionally, you may want to setup database comments if you wish to explore the database schema:
psql -U seqware seqware_meta_db < comments_on_tables.sql